
When making tough decisions, humans have long sought advice from a higher power.
In ancient times, it was oracles such as Pythia at Delphi. Then the enlightenment and the industrial revolution gave rise to disciplinary experts. In the digital age, algorithms and computers have become trusted advisors.
But the role of experts has recently come under question. Government bureaucrats are often not perceived as trustworthy. Scientists may disagree about the best course of action. Computer algorithms have come under fire for perpetuating societal stereotypes and biases.
To address this, we – in partnership with universities in Australia, Finland, and Japan – developed a crowd-powered system to provide “decision support” for complex problems. We’re calling it “AnswerBot.”
Not all questions are the same
Consider the following two questions:
What car do the Ghostbusters drive?
Which car should I buy?
The first question is factual. The internet is great at answering such questions. Post a question like this on Facebook, Quora, or even Google it, and you’re likely to get an answer in moments.
The second question requires decision support, which, in the past, has been provided by experts.
Similar questions include:
Which diet should I follow?
How can I treat my back pain?
How can we deal with racism?
The internet is terrible at answering such questions. Forum-based platforms are likely to lead to heated debate at best, or a long list of random responses that are hard to interpret. Platforms like Reddit and Quora use voting to float “good answers” to the top. However, the answers might be biased, incomplete, or fail to consider the perspective of the person asking the question.
Tapping into the wisdom of the crowd
In 1906, the English statistician Sir Francis Galton observed a competition in the UK town of Plymouth, where the aim was to correctly guess the weight of a cow. The median guess of 800 participants was accurate within 1% of the correct answer. Indeed, it was more accurate than any single person – even the cow experts. This finding came to be known as the “wisdom of the crowd”.
Our tool seeks to answer questions that require decision support by tapping into the “wisdom of the crowd”. The approach is simple, yet has already produced promising results. Here’s how it works:
Step 1
Work out what question needs to be answered. It has to be formulated in a manner that requires decision support. Let’s assume the question is “Which car should I buy?”
Step 2
AnswerBot begins posing this question to human contributors. It asks them to write down possible solutions, such as Mercedes, BMW, Mazda or Ford.
Step 3
The system asks the contributors to include criteria that should be considered, such as safety, price, fuel consumption, acceleration and comfort.
Step 4
The system generates all the possible solution-criteria combinations from the previous two steps and asks the contributors: “On a scale 0-100, how would you rate [Mercedes] in terms of [safety]”? By iterating through all combinations of cars and criteria, we construct a knowledge base that captures the collective wisdom of hundreds of people.
Finally, our website displays various criteria (safety, price and so on). Users can tweak associated sliders based on their personal preferences, and the system recommends a set of answers, ordered by their suitability.
Effectively, our system solves an optimisation problem – we use machine learning to rank the most suitable answers in our knowledge base, given someone’s preferences.
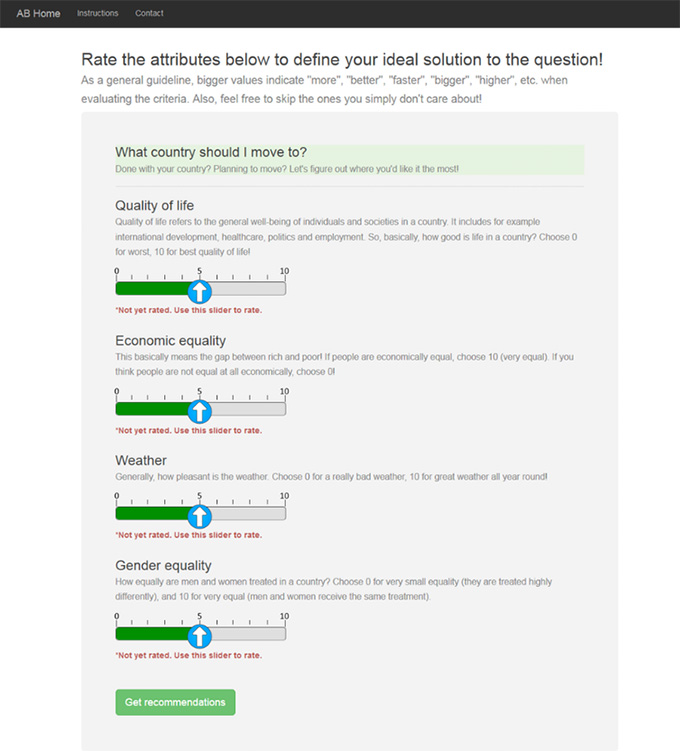
AnswerBot. Author provided
As problems become more challenging, solutions become riddled with trade offs. A person may trade off fuel efficiency to get a car that has a better safety rating. But inability to communicate these trade offs can lead to lack of trust in the decision and decision maker, or to any recommendations that one receives.
Our tool makes transparent the complex trade offs required decision making by involving a large number of people in the process.
Our approach can work even with small numbers
AnswerBot has been deployed to answer a number of “small” and “large” problems, ranging from finding a restaurant or movie, to choosing a weight loss diet, treating lower back pain, and dealing with racism. We have found that it is a great way to summarise a heated debate with hundreds of participants – and it is also possible to filter out “noise” or inappropriate answers.
By partnering with organisations and online media we can attract hundreds of contributors to build the knowledge base – although we have shown that our approach can work with just tens of contributors if there is strong agreement among them.
And our evaluations have found the quality of generated solutions are comparable to traditional online platforms like IMDB and TripAdvisor, and its recommendations are better than just randomly choosing one of solutions.
AnswerBot can also be used to test perceptions of the crowd. We asked it to answer the question “How can I treat my back pain”?
After collecting our knowledge base from hundreds of people, we showed our results to medical experts and gave them the opportunity to interact with the final decision support sliders. In fact, they disagreed with some of the recommendations – for example, the crowd seemed to think that surgery is a silver bullet solution to back pain. The experts were fascinated that it revealed how patients think about back pain, and what their misconceptions are.
The wisdom of multiple experts
We also tried AnswerBot on medical experts themselves.
We conducted a study where the “crowd” was actually made up of professional back pain experts. When our results came back, we found that the experts, in fact, disagreed among themselves.
Our decision support tool captured, in a very transparent way, the collective understanding of the experts. Any single expert could now use the tool to compare their perception against their fellow experts.
Our system combines human intuition with machine learning to provide decision support. By enabling the manipulation of preferences, the system provides transparency in choosing solutions, and facilitates users to experiment with various trade offs before making a decision.
Combining the opinions of large numbers of people – experts and non-experts alike – can provide decision support for arbitrary problems.
Author Bio: Vassilis Kostakos is a Professor of Human Computer Interaction at the University of Melbourne